FDA 510k检测
发布时间:2025-04-27
FDA510(k)检测是美国食品药品监督管理局针对Ⅱ类医疗器械上市前审查的核心程序,要求申请方通过科学验证证明产品与已合法上市器械的“实质等同性”。检测涵盖生物相容性、电气安全、电磁兼容性(EMC)、软件验证及性能测试等关键领域,需严格遵循21CFR820质量体系规范及ASTM/ISO/IEC等国际标准。重点关注风险控制数据与临床适用性的关联证据链。
注意:因业务调整,暂不接受个人委托测试望见谅。
检测项目
FDA 510(k)检测体系包含六大核心模块:
生物相容性测试:依据ISO 10993系列标准评估材料与人体组织的相互作用,包括细胞毒性(USP <87>)、致敏性(GPMT/Buehler试验)、皮内反应(ISO 10993-10)及亚慢性毒性(ISO 10993-11)等子项
电气安全验证:执行IEC 60601-1全项测试(漏电流、介电强度、机械危险防护),重点验证ME设备在单一故障状态下的安全性
电磁兼容性(EMC)测试:满足CISPR 11/EN 55011辐射发射限值及IEC 61000-4系列抗扰度标准(ESD±15kV接触放电、射频场3V/m抗扰度)
软件验证与确认:按照IEC 62304实施软件生命周期管理(SLC),包括需求可追溯矩阵(RTM)、静态代码分析(MISRA C)及故障树分析(FTA)
机械性能测试:依据产品特性开展疲劳试验(ASTM F2077)、轴向压缩试验(ASTM F2267)及扭转强度验证(ASTM F543)
灭菌验证:针对环氧乙烷灭菌器械执行ANSI/AAMI/ISO 11135确认(BI挑战试验、半周期法),辐射灭菌需符合ISO 11137剂量审核要求
检测范围
510(k)检测适用于21 CFR 862-892定义的Ⅱ类医疗器械:
体外诊断设备:血糖仪(CLIA waived)、免疫分析仪(21 CFR 862.1165)、血气分析系统(21 CFR 862.1120)
有源治疗设备:高频电刀(21 CFR 878.4400)、输液泵(21 CFR 880.5725)、呼吸机(21 CFR 868.5895)
植入器械组件:骨科螺钉(21 CFR 888.3020)、心脏起搏电极(21 CFR 870.3610)、人工关节涂层材料
医用影像设备:超声诊断仪(21 CFR 892.1550)、数字X射线系统(21 CFR 892.1650)
软件即医疗设备(SaMD):AI辅助诊断软件(21 CFR 892.2050)、远程监护系统(21 CFR 880.2700)
检测方法
具体实施流程遵循GHTF/SG5/N1:2012过程确认指南:
等效器械比对法:通过FDA产品分类数据库识别predicate device(K号产品),建立对比矩阵表比较技术参数与预期用途差异
风险分析法:依据ISO 14971:2019完成危害分析报告(HAR),采用FMEA工具量化剩余风险可接受准则(ALARP原则)
统计验证法:对性能测试数据应用ANOVA方差分析(α=0.05),样本量计算基于二项式置信区间法(95%CI下限≥90%)
加速老化试验法:按ASTM F1980进行实时/加速老化模拟(Arrhenius模型),Q10系数取2.0验证有效期声明合理性
临床评估法:通过MEDDEV 2.7/1 Rev.4建立临床证据体系,包括等同器械文献综述或前瞻性临床研究方案(PSP)
检测仪器
关键检测设备需满足NIST可追溯性要求:
生物安全测试平台:CO₂细胞培养箱(Thermo Scientific Heracell VIOS)、流式细胞仪(BD FACSCanto II)、溶血指数分析系统(HemoScan HSI-1000)
电气安全分析系统
EMC测试系统
机械性能试验机
灭菌验证设备
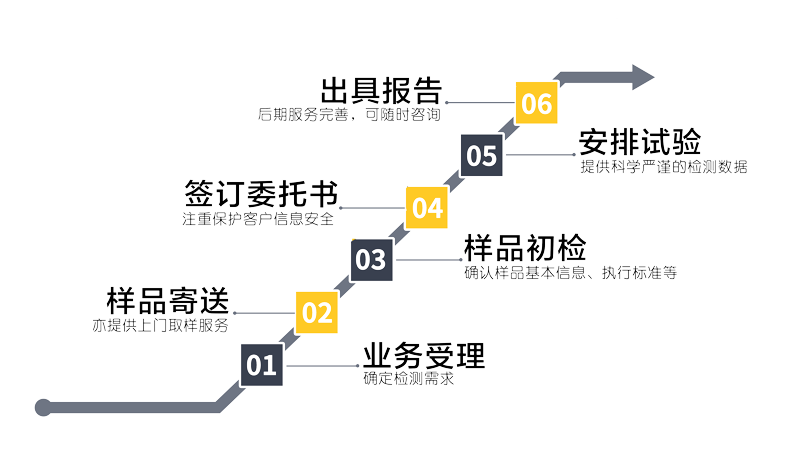
检测服务范围
1、指标检测:按国标、行标及其他规范方法检测
2、仪器共享:按仪器规范或用户提供的规范检测
3、主成分分析:对含量高的组分或你所规定的某种组分进行5~7天检测。
4,样品前处理:对产品进行预处理后,进行样品前处理,包括样品的采集与保存,样品的提取与分离,样品的鉴定以及样品的初步分析,通过逆向剖析确定原料化学名称及含量等共10个步骤;
5、深度分析:根据成分分析对采购的原料标准品做准确的定性定量检测,然后给出参考工艺及原料的推荐。最后对产品的质量控制及生产过程中出现问题及时解决。
合作客户展示

部分资质展示
